
Ahora, un esfuerzo internacional de base ha resuelto esas bases difíciles de leer, produciendo el genoma humano más completo hasta el momento. “No creo que hubiéramos podido imaginar esto incluso hace 5 años, ciertamente no hace 10 años”, dice el bioinformatico Ewan Birney, subdirector del Laboratorio Europeo de Biología Molecular y parte del Proyecto Genoma Humano original.
When it comes to sequencing the human genome, “complete” has always been a relative term. The first one, deciphered 20 years ago, included most of the regions that code for proteins but left about 200 million bases of DNA—8% of the human genome—untouched. Even as additional genomes were “finished,” some stretches remained out of reach, because repetitive segments of DNA confounded the sequencing technologies of the time. Now, an international grassroots effort has sorted out those hard-to-read bases, producing the most complete human genome yet.
In six papers in Science, the Telomere-to-Telomere (T2T) Consortium—named for the chromosomes’ end caps—fills in all but five of the hundreds of remaining problem spots, leaving just 10 million bases and the Y chromosome only roughly known. And today, the T2T consortium announced in a tweet it had deposited a correct sequence assembly of the missing Y.
“I don’t think we could have imagined this even 5 years ago, certainly not 10 years ago,” says bioinformaticist Ewan Birney, deputy director of the European Molecular Biology Laboratory and part of the original Human Genome Project “It’s a tour de force.” T2T researchers say the newly sequenced stretches reveal hotspots for gene evolution and underscore the chaotic history of the human genome. It “really gives us some insight into regions of the genome that have been invisible,” says Deanna Church, a genomicist at Inscripta, a gene-editing company.
The previously indecipherable sequences of the genome that have now come into clear view include the protective telomeres and the dense knobs called centromeres, which typically reside in the middle of each chromosome and help orchestrate its replication. Also almost completely revealed are the short arms of the five chromosomes where centromeres are skewed toward one end. Those short arms were known to contain scores of genes coding for the backbone of ribosomes, the cell’s protein factories.
When Birney, Church, and their colleagues introduced that first draft of a human genome in 2001, and even after they “completed” and published it in 2004, sequencer machines and genome assembly software could not wade through areas where the DNA sequence contained very repetitive stretches of bases: The repeats could too easily be skipped or their bases linked together incorrectly. As sequencing technology got better and costs dropped, scientists reduced the number of gaps or misassembled sequences, culminating in 2017 with the release of a human genome called GRCh38. With less than 1000 gaps, it became for many the “reference” against which other human genomes are compared.
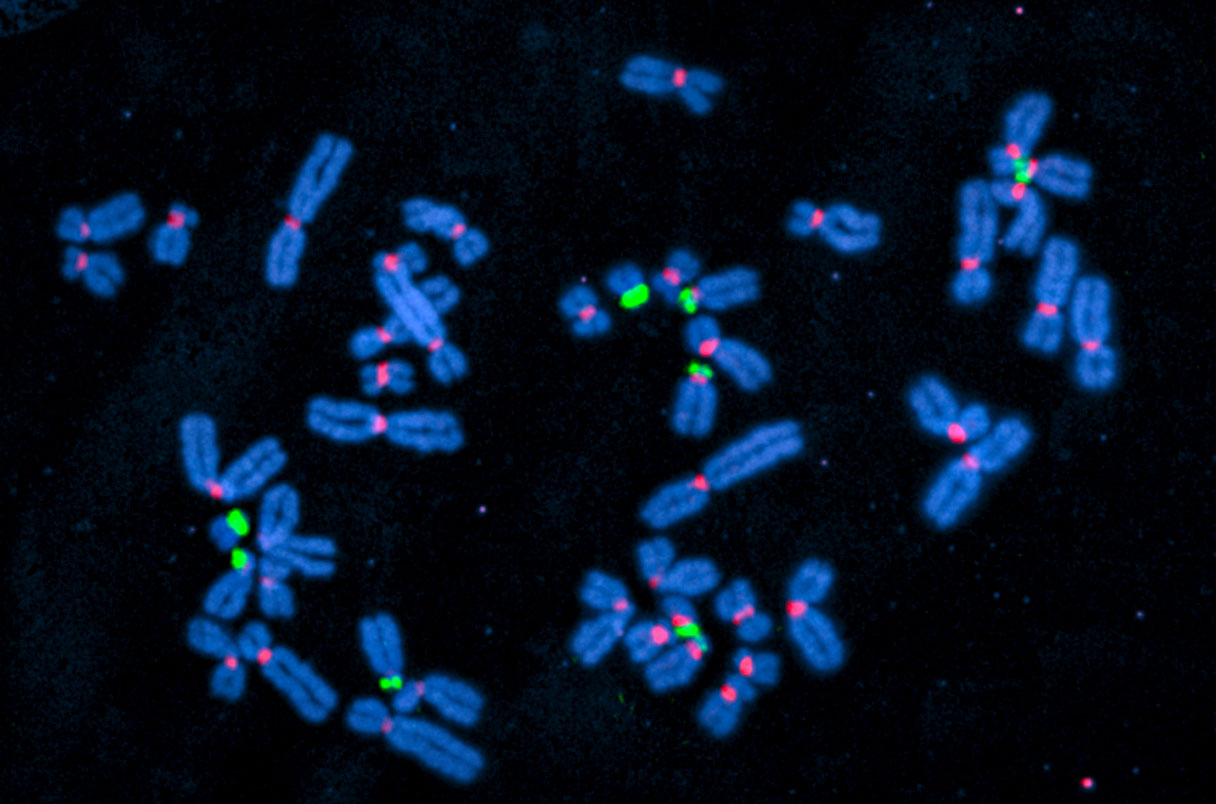
But Karen Miga and Adam Phillippy wanted to do better. Miga, a geneticist at the University of California, Santa Cruz, yearned to learn the exact sequences of the distinctive “satellite” DNA that helps form centromeres. Meanwhile, Phillippy, a bioinformatician at the National Human Genome Research Institute, was busy harnessing new sequencing technologies that could read very long stretches of DNA, reducing the need to piece together shorter sequences. After meeting at a conference, they joined forces. Then in 2019, Phillippy reported they had succeeded in sequencing the X chromosome from end to end, inspiring dozens of other researchers to join the cause. “It really took on a life of its own,” Miga says.
To simplify the task, they decided to use an anonymized cell line that was derived more than 20 years ago from an unusual growth excised from the uterus of a woman—a failed pregnancy called a mole, produced when a sperm entered an egg that lacked its own set of chromosomes. With just the sperm’s genetic material, such eggs can’t develop into an embryo, but they can still replicate, especially if the sperm delivers an X instead of Y chromosome. In a boon for the project, both members of the resulting cell line’s 23 pairs of chromosomes are identical. That “made a big difference” for eliminating gaps because sequencers didn’t have to resolve differences between the parents’ chromosomes, says Robert Waterston, a geneticist at the University of Washington, Seattle, who helped lead the Human Genome Project.
The T2T group combined sequencing technologies, including a so-called nanopore device that could read 100,000 bases at a time and another sequencer that was more accurate but only did about 10,000 bases at once. A final improvement to the latter method boosted accuracy, and together the three approaches were able to polish off all but five of the final trouble spots. “Just seeing the multiple ways they went after this [shows] these are really hard problems,” Waterston says.
The approximately 200 million bases finally in the right order and in the right place include more than 1900 genes, most of them copies of known genes. The researchers cataloged duplicated regions and mobile elements—genetic material from viruses that became incorporated into the genome. In sequencing each centromere, they learned the duplicated regions vary greatly in size, unexpected because these knobs serve the same purpose in each chromosome.
The short chromosome arms held another surprise. As expected, they included multiple copies, 400 in all, of the genes coding for the RNA that’s used to make ribosomes. “This rDNA was the last domino to fall,” as it was the hardest to sequence, Miga says.
The short arms are also “just chock-full of [other] repeats,” says Jennifer Gerton, a chromosome biologist at the Stowers Institute for Medical Research. Those include mobile elements, duplicated segments and other types of repetitive DNA, as well as many copies of genes from other parts of the genome. “It’s amazing how dynamic the human genome can be,” Church says. In five spots along these chromosomes, the resulting jumble is so long that the researchers still can’t clearly determine the order of the bases, although they have a rough idea of the sequence, Gerton says.
Short arms are likely hotspots for gene evolution, Phillippy notes, as gene copies parked there are free to mutate and take on new functions. The catalog of duplications could also shed light on neurological and developmental disorders, which have been linked to variations in the number of copies of specific sequences. Chemical modifications to the DNA in the complex repetitive areas likely play a role in disease as well, and those changes have been mapped. Because the cell line used lacked a Y chromosome, the T2T group sequenced one from a well-studied genome belonging to Harvard University systems biologist Leonid Peshkin (see sidebar, below).
Despite their latest milestone, human genome sequencers aren’t packing their bags. “There’s still some work to do,” says Human Genome Project co-leader Richard Gibbs, a geneticist at Baylor College of Medicine. He and other researchers stress that the field now needs to get similarly complete genome sequences from a greater diversity of people to look for variation in the short arms and the other tough-to-read regions, which could play a role in diseases or traits.
The T2T team has made a start by deciphering 70 more genomes, with a goal of 350 from people of diverse ancestries. These genomes, sequenced as part of the Human Pangenome Reference Consortium, are more challenging to finish because they don’t have identical pairs of chromosomes. So, for now, the team has settled for high-quality genomes that place as many of the bases as possible on their correct chromosomes. Next, the researchers plan to apply all their methods to Peshkin’s whole genome. And, eventually, Phillippy says, “We want every genome to be telomere to telomere.”
Fuente: https://www.science.org